Филадельфийская хромосома (Рh'-хромосома) - цитогенетическая аномалия, характеризующаяся нарушением в локусе 22q11. Чаще всего она обнаруживается в виде реципрокной транслокации t(9;22)(q34;q11), но может быть представлена и делецией. Впервые данное нарушение кариотипа было описано при хроническом миелолейкозе (ХМЛ) Nowell и Hungerford в 1960 году. Результатом этой транслокации является возникновение двух новых генов: гена BCR-ABL на 22 (Ph)-хромосоме и гена ABL-BCR на производной от хромосомы 9.
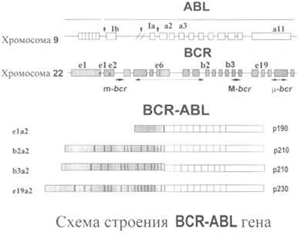
В настоящее время установлено, что основная роль в развитии патологических проявлений ХМЛ связана с геном BCR-ABL; функциональная роль гена ABL-BCR остается неясной. В зависимости от точки разрыва гена BCR могут возникать три основных типа гена BCR-ABL (рис. 1). Основной гибридный ген, характерный для классического миелолейкоза, возникает при разрыве в области M-bcr (major breakpoint cluster region). Считывание этого гена приводит к образованию двух типов химерной мРНК: b2a2 или b3a2. В итоге этой генной перестройки образуется цитоплазматический белок (p210BCR-ABL) с молекулярной массой 210 kDa, который является онкопротеином, ответственным за большинство, если не за все, фенотипические аномалии, возникающие в хронической фазе (ХФ) ХМЛ. Значительно более редко при ХМЛ встречаются другие гибридные гены, при которых точка разрыва возникает в области m(minor)-bcr (е1а2 транскрипт, белок р190) или в зоне (micro)-bcr (e19a2 транскрипт, белок р230). Заболевание в этих случаях может иметь специфический фенотип (моноцитоз при выработке белка р190) или нейтрофильно-тромбоцитарные компоненты. Гибридный BCR-ABL белок р210 обладает повышенной тирозинкиназной активностью в сравнении с белком c-Abl, образуемым в отсутствие мутации. Белок р210 локализован в цитоплазме, в то время как нормальный белок p145 расположен в ядре. За счет своего аномального положения и повышенной тирозинкиназной активности он трансформирует стволовые клетки в клетки ХМЛ, причем все трансформирующие функции белка Bcr-Abl зависят от тирозинкиназной активности Abl-фрагмента этого белка. Этот протоонкоген в норме управляет образованием ферментного белка тирозинкиназы, в то время как мутантный комбинированный ген управляет синтезом аномально длинного белка тирозинкиназы, который вызывает тяжелое нарушение контроля размножения и продолжительности жизни пораженных лейкозных клеток. В норме созревание и пролиферация кроветворных клеток находятся под контролем костномозгового стромального микроокружения, которое регулирует их взаимодействие с помощью ростовых факторов. С наличием белка p210 в лейкемических клетках-предшественницах связывают нарушение проведения сигналов, обеспечивающих нормальное функционирование клетки. Вмешательство в основные клеточные процессы приводит к злокачественной трансформации клеток и нарушению таких процессов, как контроль над пролиферацией, адгезией и апоптозом. Подтверждением ведущей роли p210 в возникновении заболевания служат эксперименты на мышах, у которых после трансфекции им клеток, содержащих ген BCR-ABL, возникало заболевание, напоминающее ХМЛ.
Ph-хромосома обнаруживается у 95% больных с типичной клинической картиной ХМЛ; почти всегда её молекулярным последствием является образование белка р210. При использовании современных методов лечения - трансплантации стволовых клеток, применении гидроксимочевины, интерферона-α (ИФНα) – излечение наступает менее чем у 20% пациентов с ХМЛ, хотя для перестройки типа в3а2 считается благоприятним применение INF-α. Выявление белка р 210 в периферической крови или костном мозге через 6 – 36 месяцев после аллогенной ТКМ ассоциировано с высоким риском рецидива.
Кроме того, транслокация t (9; 22) (q34; q11) выявляется у 10–20% взрослых и 2–5% детей, больных острым лимфобластным лейкозом (ОЛЛ), причем у большинства детей и примерно половины взрослых больных она приводит к синтезу белка р190, редко обнаруживаемому при ХМЛ. Ph+ ОЛЛ имеет плохой прогноз, выживаемость составляет менее 20% у взрослых и 25-30% у детей. Чаще это дети старшего возраста, с гиперлейкоцитозом, ОЛЛ В-линейного фенотипа. Выход в ремиссию может произойти, так же как и у Рh- ОЛЛ, но затем возникает ранний рецидив. Дети младше 10 лет, с лейкоцитозом менее 25 тыс. в 1 мкл и хорошим ответом на 7-й день лечения имеют 50-процентный шанс на выживаемость. Обнаружение филадельфийской хромосомы при ОЛЛ может служить фактором развития нейролейкемии. Есть данные, что одной из причин плохого прогноза является медленный ответ на терапию индукции почти у половины (46%) больных и высокая частота рецидивов. В плане лечения таких пациентов предпочтение отдается аллогенной ТКМ от совместимого родственного донора в первые 6 месяцев заболевания, что снижает риск токсической смерти и рецидива. Аутотрансплантация костного мозга или трансплантация костного мозга от неродственного донора не имеют преимуществ перед интенсивной химиотерапией из-за высокой частоты токсических летальных исходов.
При ОМЛ филадельфийская хромосома встречается у 2 – 6 % больных и также ассоциирована с плохим прогнозом.
Исходя из общепринятой концепции о связи гена BCR-ABL и ассоциированных с ним белков p190 и p210 с повышенной тирозинкиназной активностью и развитием лейкоза, логичным является разработка терапевтических направлений, в которых этот ген представляет мишень для целенаправленного воздействия, т. е., можно с уверенностью предсказать, что ингибитор Abl-тирозинкиназы будет эффективным и избирательным средством лечения ХМЛ и других Bcr-Abl-положительных лейкозов. Препарат STI571, избирательно ингибирующий Bcr-Abl-тирозинкиназу, представляет собой специально сконструированный ингибитор Abl-тирозинкиназ, рецепторов фактора роста тромбоцитов и c-kit-тирозинкиназ. У больных с ХМЛ он дает очень хороший терапевтический эффект при минимальных побочных эффектах.
Классический случай транслокации с образованием химерного гена BCR/ABL обнаруживается стандартным цитогенетическим анализом, но в 5-7% случаев транслокация встречается на субмикроскопическом уровне, которая может быть обнаруженной лишь при FISH-анализе или с применением ПЦР.
Набор реактивов ОНКОСКРИН-1-1 предназначен для выявления экспрессии онкогена BCR/ABL типов в3а3 и в2а2 у больных ХМЛ, ОЛЛ и ОМЛ. Анализируемым материалом является периферическая кровь и костный мозг. Данная тест-система основана на методе обратной транскрипции-полимеразной цепной реакции (ОТ-ПЦР) с использованием праймеров, комплементарных области слияния bcr и abl последовательностей в составе химерной мРНК BCR/ABL. Эта мРНК при помощи обратной транскриптазы переводится в ДНК-копию (кДНК), которая служит матрицей для Taq-полимеразы. При участии этого фермента в процессе ПЦР осуществляется многократный синтез фрагментов ДНК, содержащих точку разрыва (слияния) BCR и ABL последовательностей. Дополнительный раунд ПЦР с применением праймеров, сдвинутых внутрь BCR/ABL-матрицы («гнездовая ПЦР»), повышает чувствительность анализа. Данная тест-система выявляет в положительных образцах BCR/ABL-фрагменты ДНК, которые имеют строго определенный размер и электрофоретическую подвижность, благодаря чему их легко отличить как от отрицательных образцов, так и от образцов, в которых обнаружены неспецифические фрагменты, не имеющие диагностического значения.
Источник: http://newdiagnostics.dn.ua/index.php?option=com_content&task=view&id=21&Itemid=29&&lang=ru